| HOME | RESEARCH | TEACHING | PUBLICATIONS | SPONSORS | CONTACT |
|---|
Research
Interfacial Phenomena
Molecular beam studies have shown that the thermalization of molecular
species impinging on liquid surfaces is highly efficient. The higher efficiency
 observed for simple hydrocarbons as compared to hydrogen bond-forming liquids
demonstrates how the corrugation of liquid interfaces depends strongly on the
temperature of the liquid and the rigidity of the bulk phase.
Because the diffusivity of the liquid phase, computer simulations of liquid
interfaces are technically difficult, and some of the technical
details implemented in the calculations can compromise the physical
meaning of the results.
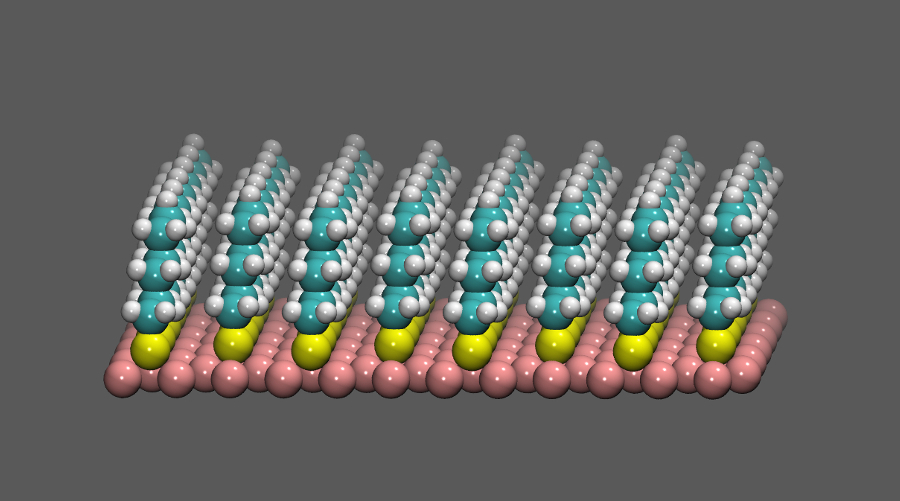
The picture in the right shows a self-assembled monolayer
(SAM) made of six-carbon thiol molecules.
Self-assembled monolayers (SAM) are highly ordered, crystal-like, oriented monolayers
of long-chain functionalized molecules standing self-supported on a metallic surface.
observed for simple hydrocarbons as compared to hydrogen bond-forming liquids
demonstrates how the corrugation of liquid interfaces depends strongly on the
temperature of the liquid and the rigidity of the bulk phase.
Because the diffusivity of the liquid phase, computer simulations of liquid
interfaces are technically difficult, and some of the technical
details implemented in the calculations can compromise the physical
meaning of the results.
The picture in the right shows a self-assembled monolayer
(SAM) made of six-carbon thiol molecules.
Self-assembled monolayers (SAM) are highly ordered, crystal-like, oriented monolayers
of long-chain functionalized molecules standing self-supported on a metallic surface.
 Since the molecules are anchored by only one end, they retain some of the mobility that
they have in the liquid but lose all the fluidity of the liquid
phase. We have used self-assembled monolayers to mimic the liquid interface.
The corrugation of the interface can be tuned by varying the length of the
monolayer molecules and its temperature.
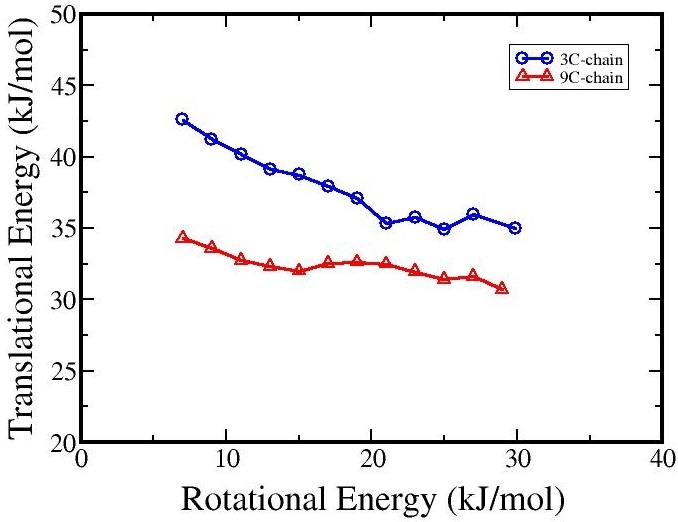
Figure in the left shows the rotational excitation for NO scattering from two monolayers of
different thickness. Longer monolayer molecules posses greater mobility which confers
a higher corrugation to the interface. The higher corrugated surface promotes more
rotational excitation. As demonstrated by the flatter curve for the monolayer with
the nine-carbon chain molecules.
Since the molecules are anchored by only one end, they retain some of the mobility that
they have in the liquid but lose all the fluidity of the liquid
phase. We have used self-assembled monolayers to mimic the liquid interface.
The corrugation of the interface can be tuned by varying the length of the
monolayer molecules and its temperature.
Figure in the left shows the rotational excitation for NO scattering from two monolayers of
different thickness. Longer monolayer molecules posses greater mobility which confers
a higher corrugation to the interface. The higher corrugated surface promotes more
rotational excitation. As demonstrated by the flatter curve for the monolayer with
the nine-carbon chain molecules.
Heat Transfer
The phenomenological coefficients describing the heat transfer and other
transport properties can be obtained from equilibrium time-correlation
functions. This approach, however, is computationally expensive since it
requires simulation times several times longer than the characteristic
relaxation time of the correlation itself.
We have used Non-Equilibrium Molecular Dynamics (NEMD) to perform computational
experiments that provide a phenomenological description of the transport process.
This approach is particularly suitable for complex systems (i.e. molecular liquids
and solids) since molecular dynamics simulations in such systems can be really
"sluggish". A test run to determine the thermal conductivity of Si was done by
placing the solid material in between two heat reservoirs with different
temperatures and the thermal coefficient was determined from the phenomenological
laws describing the process. The computational experiment began with the realization
of an thermodynamic equilibrium state at a given temperature by setting both
reservoirs at the same temperature until a constant temperature state is reached
for the whole system. Then, the temperature of one of the reservoirs was set to
a lower value and the system was allowed to relax to a stationary non-equilibrium
state with a thermal gradient between the two reservoirs.
Once the system has reached a stationary state, the local equilibrium hypothesis
is applied and the heat current is calculated in terms of properties of each
individual atom using Kirkwood's equation:
$$\boldsymbol{q}_{i}\left(\boldsymbol{r};t\right) = \left[ \boldsymbol{v}_{i}\cdot E_{i} +
\frac{1}{2}\sum_{i\neq j} \boldsymbol{r}_{ij}\cdot\left(\boldsymbol{F}_{ij}\cdot
\boldsymbol{v}_{i}\right)\right]\delta\left(\boldsymbol{r}_{i}-\boldsymbol{r}\right)$$
The heat current for the system can be calculated as the canonical ensemble average
of the time-averaged heat current for each individual layer perpendicular to the
direction of the temperature gradient.
The figure below shows the calculated and experimental thermal conductivity.
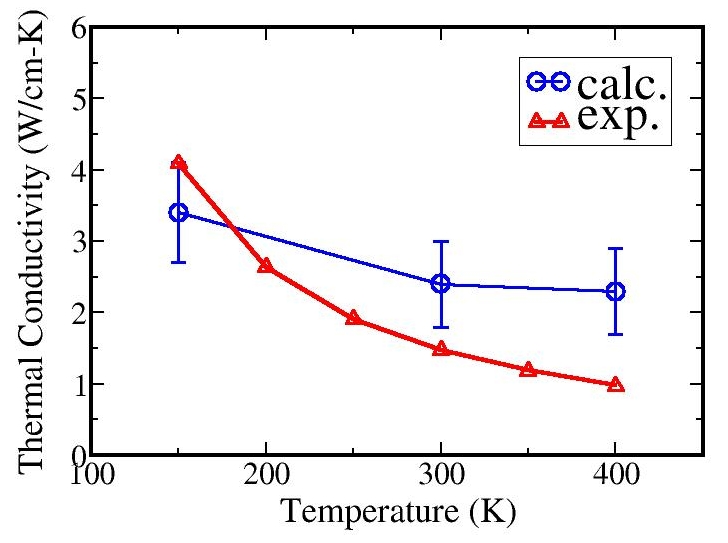 The underestimation can be corrected with a refined parameterization of the potentials.
The method describes the expected trend correctly: the thermal conductivity decreases
with increasing temperature which is due mainly to anharmonic effects in the lattice
vibration that become predominant at high temperatures.
The underestimation can be corrected with a refined parameterization of the potentials.
The method describes the expected trend correctly: the thermal conductivity decreases
with increasing temperature which is due mainly to anharmonic effects in the lattice
vibration that become predominant at high temperatures.
Surface Wetting
The wetting capabilities of the liquids have been studied simulating the morphological transformation
undergone by a drop of the liquid when going from being isolated in the vacuum to its equilibrium
state resting on top of a quartz surface. The figure below shows the equilibration process of the liquid
drop on a quartz surface:
 Once the equilibrium state was achieved the wetting capabilities of the liquid were determined by measuring
the contact angles with the surface as depicted in the figure below.
Once the equilibrium state was achieved the wetting capabilities of the liquid were determined by measuring
the contact angles with the surface as depicted in the figure below.
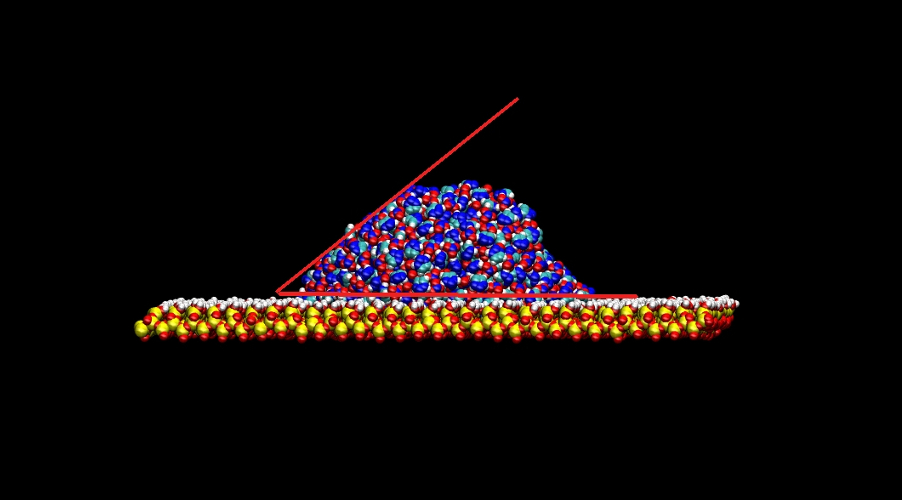 The magnitude (< 90 degrees) of the contact angles indicates that there is a favorable
solid-liquid interaction. This also suggests that these liquids may posses good lubrication properties.
The magnitude (< 90 degrees) of the contact angles indicates that there is a favorable
solid-liquid interaction. This also suggests that these liquids may posses good lubrication properties.
Tribological Properties
The tribological properties of the liquids were studied by simulating the lubrication process. The liquid
was confined between two quartz surfaces and equilibrated. Once the thermodynamic equilibrium was attained.
The lubrication process was simulated by sliding the top surface at a constant velocity while keeping the
bottom surface stationary. The animation below demonstrates the computational experiment performed.
 The top slab "drags" the upper-most layer of the liquid as it slides toward the right.
The momentum transfer to the liquid creates a velocity gradient in the direction perpendicular to the surface.
This velocity gradient is directly proportional to the liquid's viscosity.
This phenomenological approach allows for the direct determination of the viscosity of the liquids without
numerical artifacts.
The top slab "drags" the upper-most layer of the liquid as it slides toward the right.
The momentum transfer to the liquid creates a velocity gradient in the direction perpendicular to the surface.
This velocity gradient is directly proportional to the liquid's viscosity.
This phenomenological approach allows for the direct determination of the viscosity of the liquids without
numerical artifacts.